Systemic Bone Loss after Fracture
Two million broken bones accounting for $19 billion dollars in medical costs occur each year due to osteoporosis. Many factors affecting fracture risk have been identified, but the best predictor of a future fracture is a previous history of fracture at any skeletal site, even after controlling for bone mineral density. A likely contributor to this phenomenon is that a fracture causes a systemic loss of bone that, if not fully recovered, permanently compromises skeletal health. Mechanisms of this response may include systemic inflammation, bone adaptation to mechanical disuse, increased rates of bone remodeling (both local and systemic), and utilization of mineral from the skeleton for formation of a fracture callus. However, the systemic loss of bone following an initial fracture has not been fully quantified, and the specific mechanisms and age-, sex-, and hormone-dependence of this bone loss have not been identified. The long-term goal of this research is to investigate systemic loss of bone mass and strength following bone fracture, and to translate these findings to human health and treatment of clinical fractures. We hypothesize that fracture initiates a systemic loss of bone due to systemic inflammation, increased bone turnover, and decreased voluntary mechanical loading (disuse).

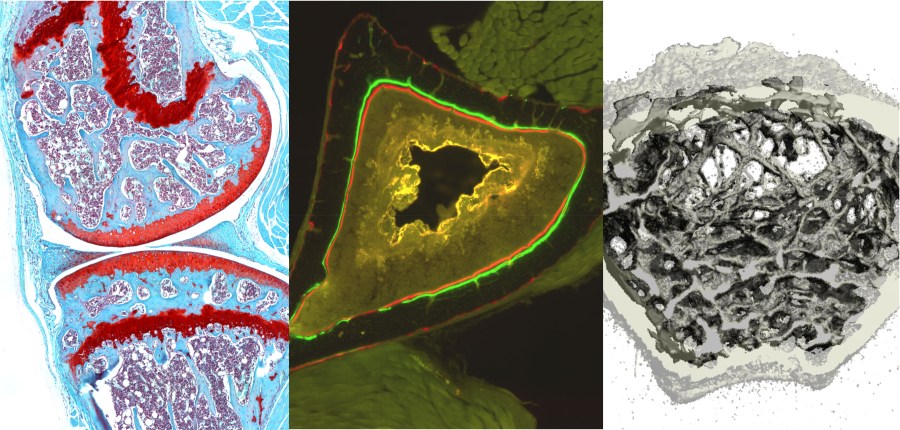
Post-Traumatic Osteoarthritis (PTOA)
Post-traumatic osteoarthritis (PTOA) is a common consequence of traumatic joint injury, with at least 50% of individuals developing PTOA within 10-20 years following an injury such as anterior cruciate ligament (ACL) rupture. Mechanisms of PTOA development likely include changes in the biomechanical stability and mechanical loading of the joint, and injury-induced biological processes including inflammation, tissue degradation, and subchondral bone and articular cartilage remodeling. However, the relative contributions of these mechanisms to PTOA development are not fully known, and the window of opportunity for treatments aimed at preventing PTOA has not been established. The long-term goal of this research is to determine the time course and mechanisms of PTOA progression, and investigate therapies that can be applied at the time of injury that will slow or prevent the onset of PTOA. In particular, we are interested in biomechanical and bone-related processes contributing to PTOA, including joint instability, early phase rest vs. mobilization, subchondral bone remodeling, and osteophyte formation. In my lab we have developed a novel non-invasive mouse knee injury model that uses a single cycle of tibial compression overload to rupture the ACL, inducing inflammation and biomechanical changes in the joint. This injury method is a significant step forward for mouse models of PTOA, since it creates a joint injury response relevant to traumatic joint injury in humans.

